RESEARCH1
RESEARCH研究内容
RESEARCH1
これまで当研究室では、近年発展の目覚ましい非平面多芳香族化合物であるヘリセン類の 「C–H結合活性化を活用した新規合成法」 と「新規多重ヘリセン類の合成、構造、および物性」について顕著な成果をあげてきた。ヘリセンの合成手法として代表的なスチルベン類の光環化反応は、高次ヘリセン合成における基質合成の煩雑さを回避できる有用な手法の1つである。しかしながら、過剰酸化を併発する点や、大希釈条件下での反応が必要とされる点において、さらなる改善の余地が残されていた。これらの問題点を解決すべく、C–H 結合活性化を伴う炭素-炭素形成反応を活用したヘリセン類の効率的な合成手法の開発に着手し、基質合成の簡便化を図るとともに、狙った位置での炭素-炭素結合生成を複数箇所で行うことに成功した。以下に具体例を述べる。
1. C–H 結合活性化を活用したヘリセン類の効率的な合成法の開発
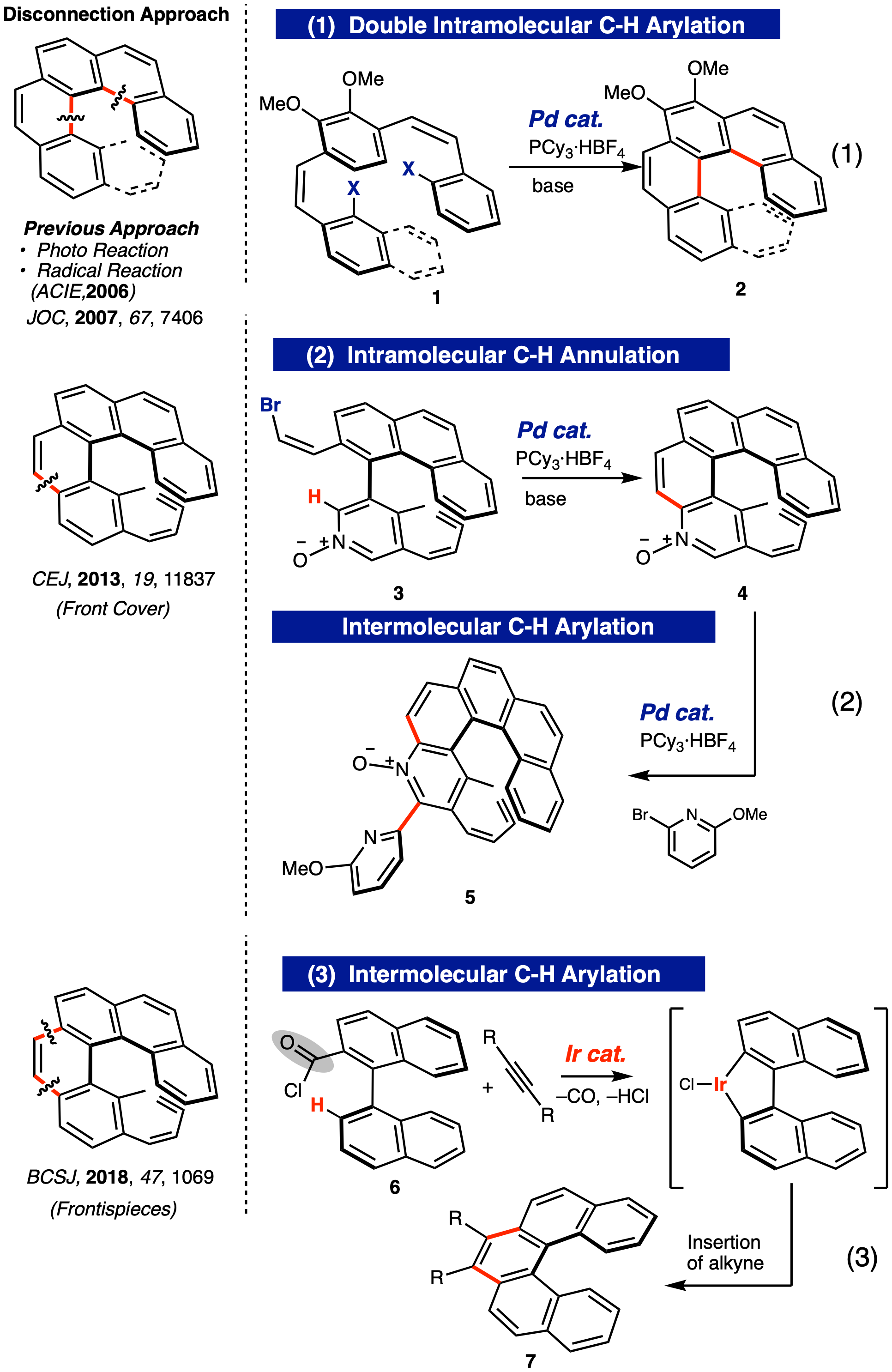
ダブル分子内C-Hアリール化によるカルボヘリセン類の合成
ビススチルベン類 1 に対するPd 触媒分子内ダブルC–Hアリール化による [5]、および [6]カルボヘリセン類 2の合成法を確立した(式 1)。本反応は、対応するビススチルベン誘導体を基質に用いた光反応による合成上の欠点を克服するとともに、C–Hアリール化を用いたヘリセン合成として最初の報告例である (JOC, 2007, 72, 7406)。
ビニルブロミド側鎖との分子内C-Hアリール化によるアザヘリセンオキシド類の合成
ヘリセン合成において最もねじれる中央の芳香環をどのように構築するかは重要な点である。我々は、ビアリール誘導体 3 の側鎖に導入したZ-ブロモビニル基を活用して、Pd触媒分子内 C–H アリール化反応により対応するアザヘリセンオキシド 4を効率よく合成する方法論を確立した(式 2)。さらに、N-オキシドのもう一方のオルト位 C-H 結合にも、Pd 触媒分子間 C–H アリール化反応により新たにアリール基を導入することに成功し、有機触媒に潜在的に利用可能なキラルヘリセン誘導体 5 の合成を達成した (CEJ, 2013, 19, 11837, Front Cover)。
分子間環化C-Hアリール化によるカルボヘリセン類の合成
式2の手法をさらに発展させて、Pd触媒では達成できなかったアルキンとビアリール誘導体 6 との分子間C–H アリール化反応を Ir 触媒を用いることで達成し、カルボヘリセン類 7 のさらなる新規合成法を確立した(式 3)(BCSJ, 2018, 39, 1069)。
脱水素型ダブルC-Hアリール化によるS字型ジアザヘリセンの合成、およびPd錯体化
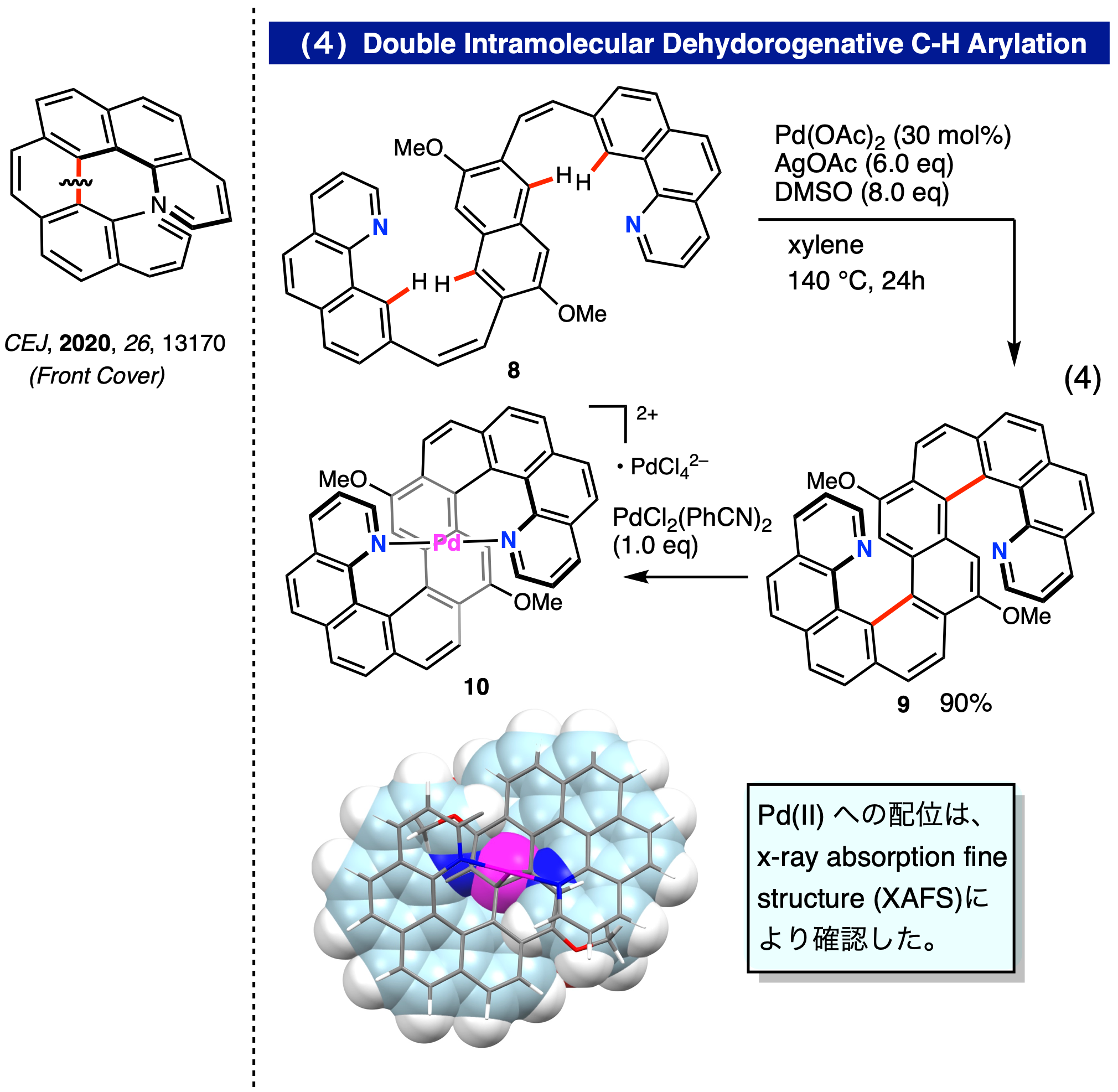
これまで基質にハロゲンなどの活性基の導入を必要としたが、ハロゲン導入を必要としない脱水素型ダブル C–H アリール化反応を行うことでS字型ジアザヘリセン 9 の合成を達成した(式 4)。一般に収率が著しく低下する高次ヘリセン合成では前例にない高収率 (90%) にてdl体 9を選択的に合成することができた。また、得られた 9 はヘリセン内縁部に2つの配位性窒素原子を持つため、トランスキレート型配位子として活用できることが期待された。実際に、S字型ジアザヘリセンとPdCl2(PhCN)2とを混合すると瞬時に Pd 錯体 10 が生成し、2つの窒素原子が Pd に対してトランス位で配位していることを x-ray absorption fine structure (XAFS) を用いて確認することに成功した。ジアザヘリセンをトランスキレート型二座配位子として活用できることを示した最初の例である (CEJ, 2020, 26, 13170, Front Cover)。
2. 多重ヘリセン類の合成、構造、および物性
現在、ヘリセンの化学がこの数年で新たなステージに突入し、現在大いに脚光を浴びている。これは分子内にヘリセン構造を複数有する「多重ヘリセン」と呼ばれる分子の開発研究が世界中で爆発的に進んでいることが要因である (。多重ヘリセンは、単独のヘリセンでは見られない通常よりも大きく湾曲した構造になる点に加えて、多様な電子物性の発現や高度な不斉環境の構築を可能にする点において、世界中の化学者の興味を惹きつけている。我々は、この多重ヘリセンブームのさきがけとなる2015年にダブルヘリセンの合成を報告し、それ以降において、この分野を牽引してきた。以下にその取組について述べる。
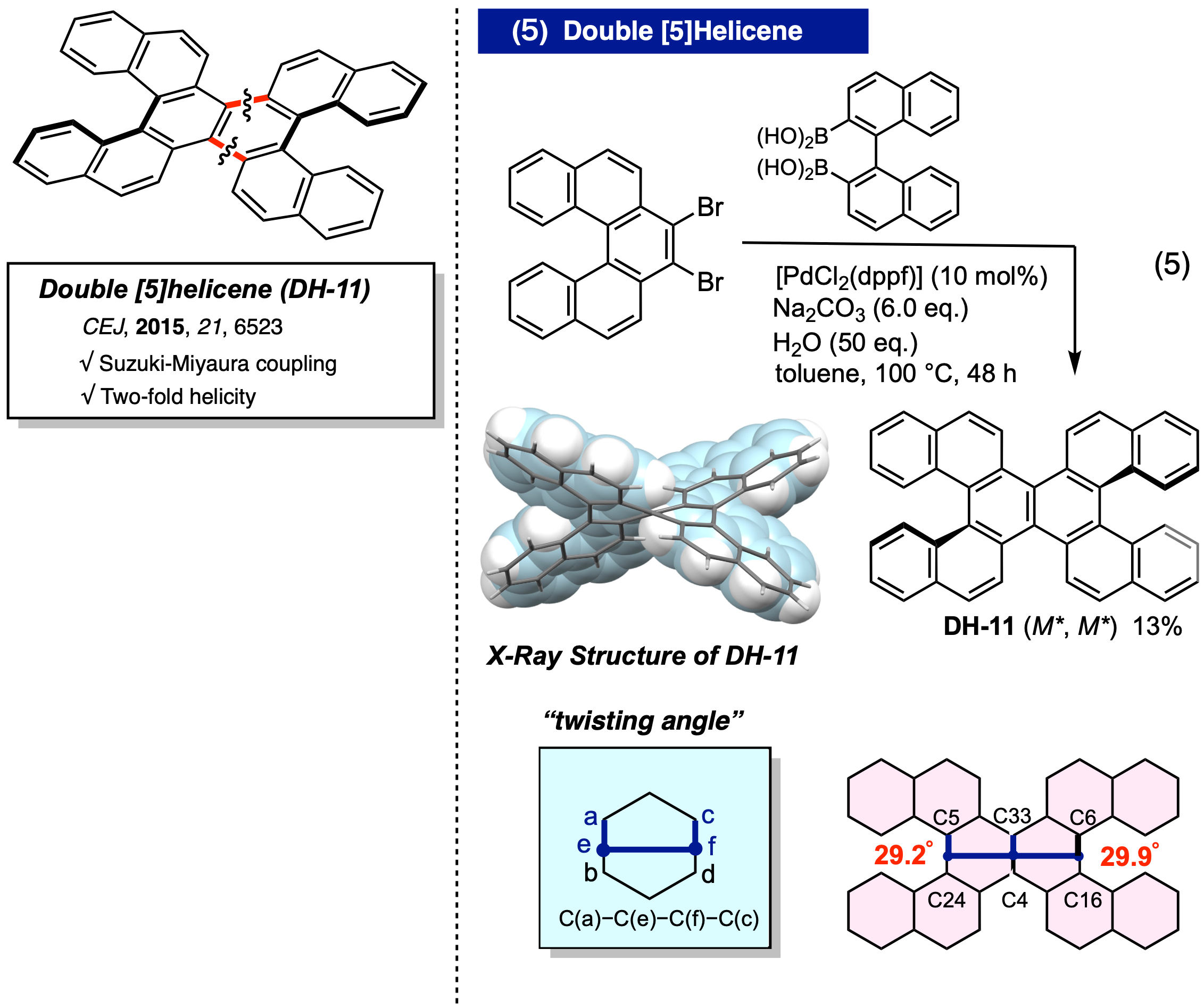
ダブル鈴木―宮浦反応によるダブルヘリセンの合成
7,8-ジブロモ[5]ヘリセンとビナフチルビスホウ酸とのダブル鈴木―宮浦カップリング反応により、dl体のダブルヘリセン (DH-11) を選択的に合成することに成功した。この際に、X線結晶解析の結果から中央のナフタレンコアにおけるベンゼン環のねじれ角 (twisting angle: 次ページの図を参照) が最大で 29.9°になることを明らかにした。単独の [5]ヘリセンのそれが 13.7°であることから、多重ヘリセン化することで複数の立体的なひずみが中心部の芳香環に集中し、大きく湾曲した構造をとることを明らかにした (CEJ, 2015, 21, 6523)。
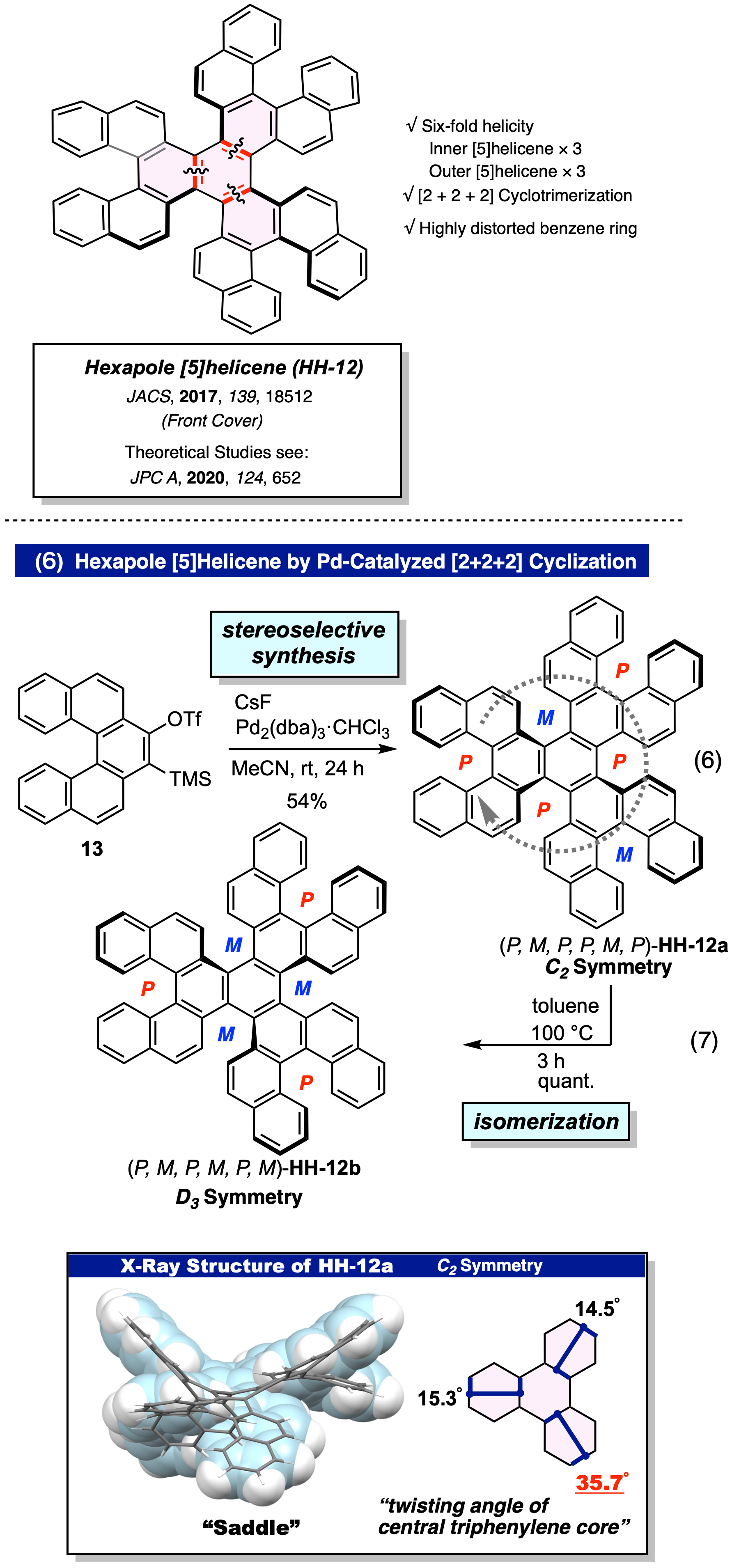
Pd触媒 [2+2+2] 環化三量化による六重ヘリセンの合成、構造、および物性
[5]ヘリニルアライン前駆体 13 を用いて中央部にアラインを発生させ、Pd 触媒による環化三量化反応を行うことで6つのヘリセン構造を内包する六重ヘリセン (HH-12) の合成に成功した (JACS, 2017, 139, 18512)。生成可能な 10種類の異性体の中から准安定な C2 対称HH-12a が立体選択的に得られた(式 6)。さらに HH-12a をトルエン中、100 ℃ で加熱することでプロペラ構造を有し、熱力学的に最安定な D3 体 (HH-12b) に変換できることも明らかにした (式 7)。
また、C2 対称 HH-12a は発表当時で最も歪んだベンゼン環 (twisting angle 35.7) を有する分子となった。さらに、HH-12a が立体選択的に得られる理由について、理論化学計算による詳細な反応機構も明らかにした ( JPC A, 2020, 124, 652)。なお、六重ヘリセンは現在報告されている単分子の多重ヘリセンの中では最も多重度の高いヘリセンである。
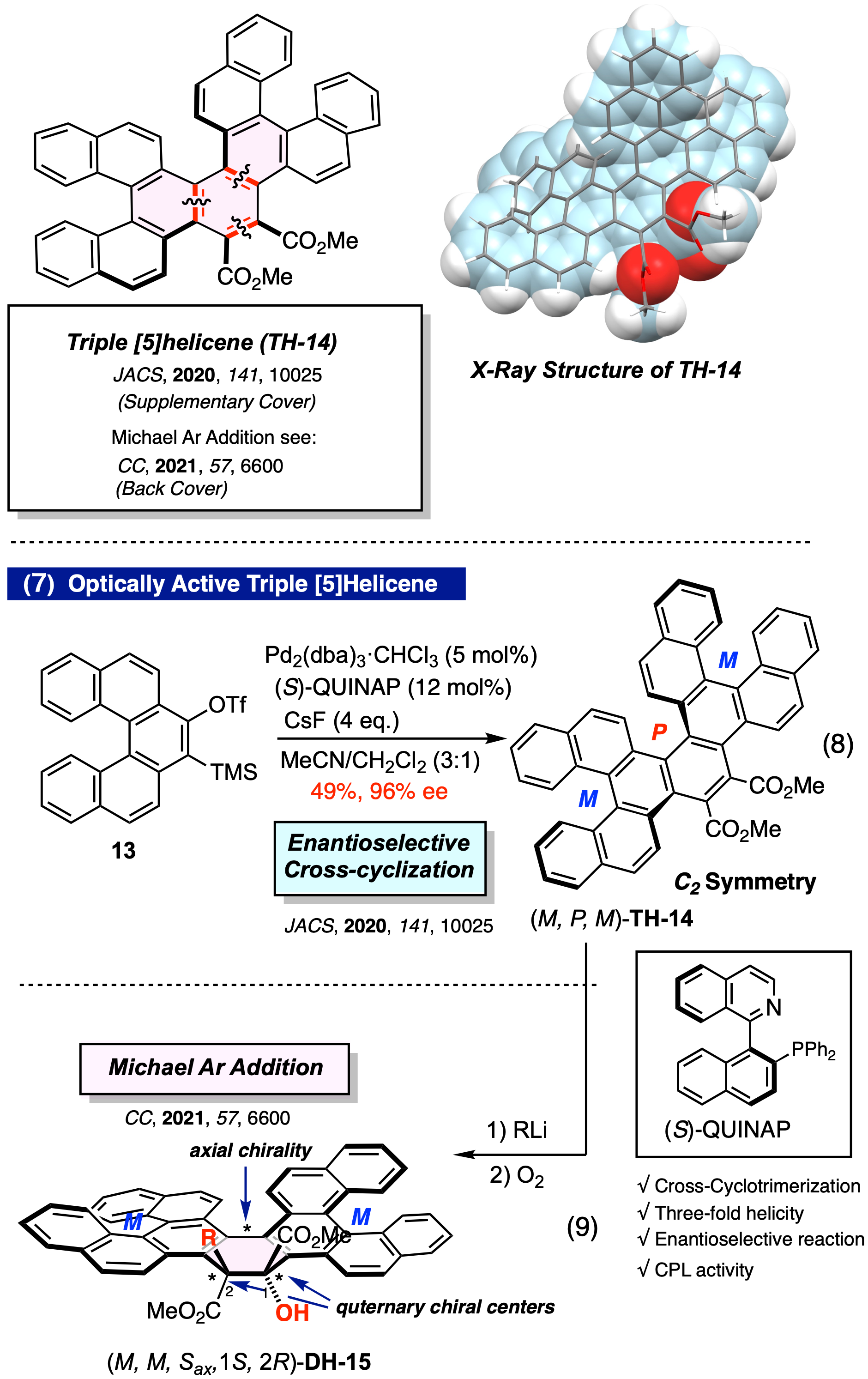
Pd触媒 [2+2+2] 不斉交差環化三量化による三重ヘリセンの触媒的エナンチオ選択的合成
これまでに合成された光学的に純粋な多重ヘリセンのほとんどは、Scholl反応などの酸化的カップリング反応後に、キラル固定相を用いた光学分割によって調製されていた。我々は、ヘリセニルアライン前駆体 13 とアセチレンジカルボン酸ジメチルとのPd触媒を用いた不斉交差 [2+2+2] 環化三量化反応による三重ヘリセンの合成を検討し、(S)-QUINAP を用いたときに収率 49%、エナンチオ選択性 96% ee で三重ヘリセン (TH-14) を光学活性体として得ることに成功した (文献4, JACS, 2020, 141, 10025)(式 8)。また、実験的、理論化学的考察を行った結果、本反応は動的速度論的分割を経て立体選択的に進行していることが明らかとなった。また、(M, P, M)-TH-14は高いCPL特性 (glum = –2.4 × 10−3 @ 421 nm) を示すことを明らかにした。
得られた三重ヘリセンのエステル基を第三級アルコールに変換し、キラルジオールとするためにメチルグリニャール試薬の付加を検討したところ、興味深いことにメチル基が芳香環上の炭素にマイケル付加し、続いて酸素分子が求電子的に捕捉されることにより、軸不斉と連続する二つの第四級炭素不斉を有するダブルヘリセン (DH-15) が得られることを明らかにした(式 9)。これは、対応するシングル[5]ヘリセンでは進行しない反応であり、多重ヘリセンのもつ特異な反応性を明らかにした最初の例である (CC, 2021, 57, 6600)。
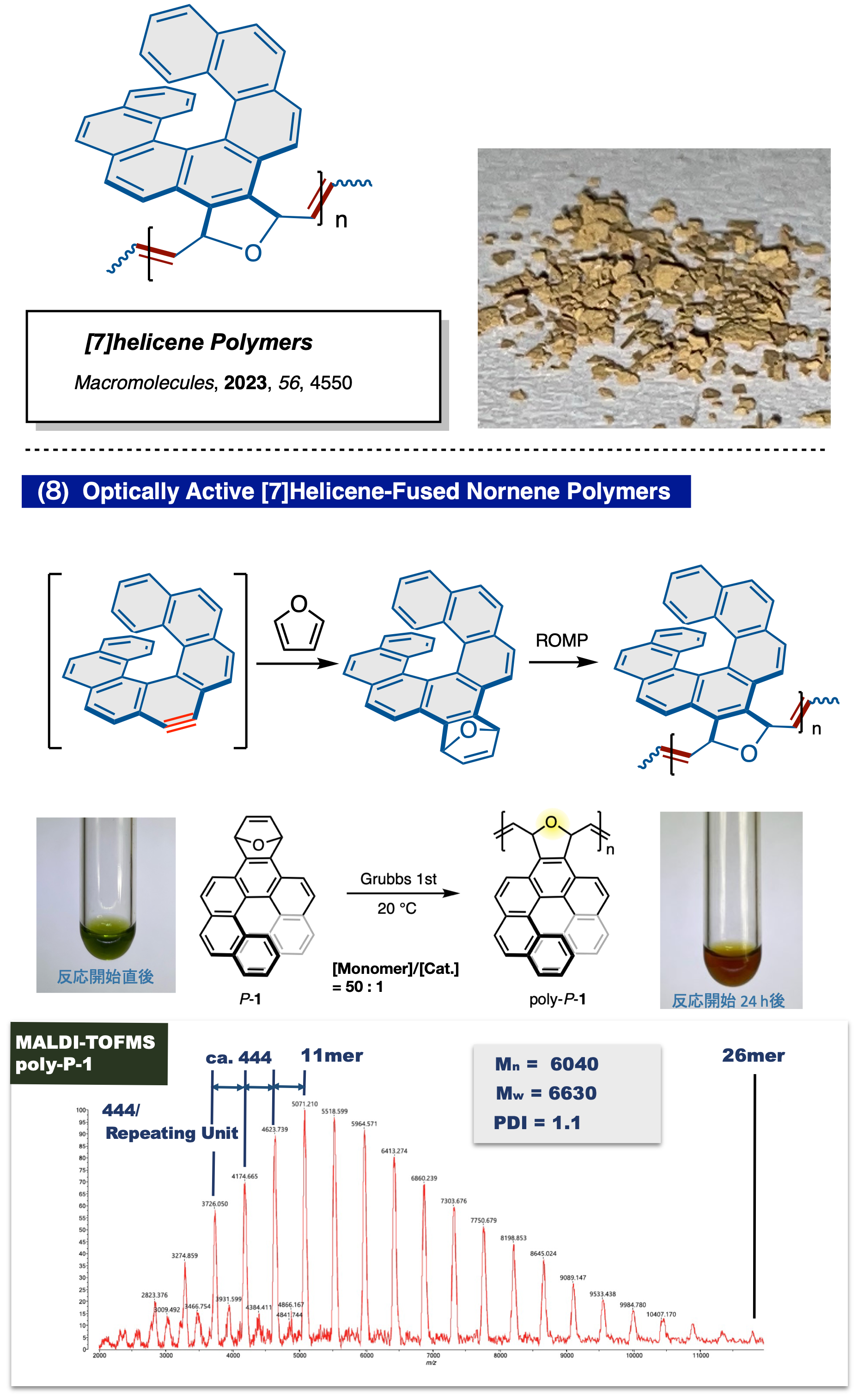
開環メタセシス重合による光学活性[7]ヘリセン融着高分子の合成
1,4-ジヒドロ-1,4-エポキシ [7] ヘリセンの開環メタセシス重合 (ROMP) を活用したポリ [7] ヘリセンの合成を行った。2,7-ジヒドロキシフェナントレンを出発物質として、9,10 位に ブロモ基を有する [7] ヘリセン誘導体を合成し、塩基存在下でアラインを発生させるとともにフランとのDiels-Alder反応を行い、モノマーを得た。次に、光学分割によりモノマーを光学活性体とし、さらにGrubbs 1st触媒による開環メタセシス重合反応 (ROMP) を行った。MALDI-TOFMSにて質量分析を行ったところ、モノマーの分子量に相当する分子イオンピークが規則的に増加するスペクトルを観測した。このことからROMP により重合し、最大で26量体となる光学活性ポリ[7]ヘリセンが得られていることを確認した (Macromolecules, 2023, 56, 4550)。