Research News
Dec 5, 2022
Quantum algorithm of the direct calculation of energy derivatives developed for molecular geometry optimization
A significant step toward social implementation of chemical quantum calculations on a quantum computer
Results of geometry optimizations for H2 molecule. Geometry optimizations with various initial values of the H–H interatomic distance revealed that the calculation quickly converges to the equilibrium bond length within 10 iterations, no matter which interatomic distance is used to start the calculation.
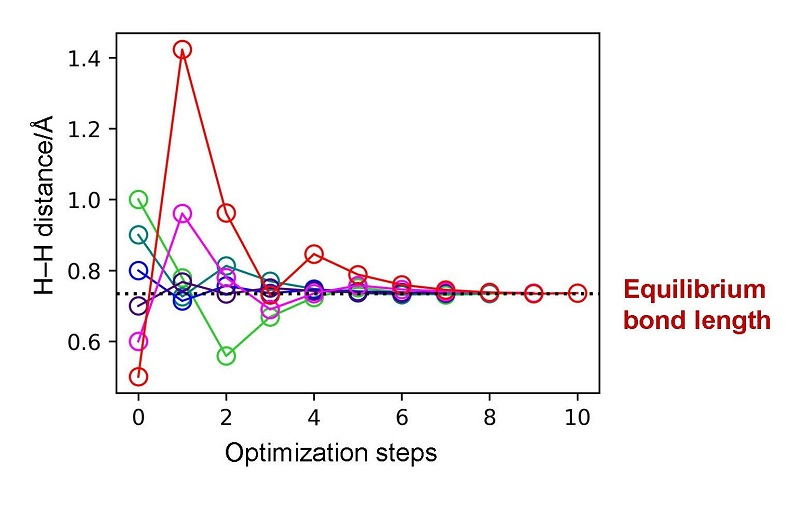
In recent years, research and development on quantum computers has made considerable progress. Quantum chemical calculations for electronic structures of atoms and molecules are attracting great attention as one of the most promising applications of quantum computers. In order to utilize quantum chemical calculations for chemistry and related fields, it is essential to develop geometry optimization methods for finding the most stable structure of molecules. The geometry optimization requires calculations of energy derivatives with respect to nuclear coordinates of molecules.
The finite difference method is one approach for energy derivative calculations. On a classical computer, calculations based on this method for one-dimensional systems require at least two evaluations of the energy. Previous research has shown that a quantum computer, in contrast, requires only a single query to calculate the energy derivatives based on the finite difference method, regardless of the number of degrees of freedom. However, quantum circuits relevant to quantum algorithms capable of performing energy derivative calculations have not been implemented.
A research group including Dr. Kenji Sugisaki, Professor Kazunobu Sato, and Professor Emeritus Takeji Takui from the Graduate School of Science at Osaka Metropolitan University has successfully extended the quantum phase difference estimation algorithm, a general quantum algorithm for the direct calculations of energy gaps, to enable the direct calculation of energy differences between two different molecular geometries. This allows for the computation, based on the finite difference method, of energy derivatives with respect to nuclear coordinates in a single calculation.
Furthermore, the research group has applied the developed energy derivative calculations to execute geometry optimizations of H2, LiH, BeH2, and N2 molecules without calculating the total energies, demonstrating the usefulness of the developed method. The group also discussed how quantum circuits can be assembled according to different degrees of freedom of the molecules.
This research is the latest in a series of the researchers’ articles on quantum chemical calculations on quantum computers. “Our latest findings bring us one step closer to applying quantum chemical calculations on a quantum computer to real-world problems,” said Dr. Sugisaki. “Since energy derivative calculations are used for not only molecular geometry optimizations but also various calculations for molecular properties, the application of our method is expected to play a very important role in a wide range of related fields, such as in silico drug discovery/design and materials development.”
Funding
This work has been supported by JST PRESTO “Quantum Software” project (Grant No. JPMJPR1914), Japan, and KAKENHI Scientific Research C (Grant No. 21K03407) from JSPS, Japan. Partial support by AOARD Scientific Project on "Molecular Spins for Quantum Technologies" (Grant FA2386-17-1-4040, 4041), USA is also acknowledged.
Paper Information
Journal: The Journal of Physical Chemistry Letters
Title: Quantum Algorithm for Numerical Energy Gradient Calculations at the Full Configuration Interaction Level of Theory
Author: Kenji Sugisaki, Hiroyuki Wakimoto, Kazuo Toyota, Kazunobu Sato, Daisuke Shiomi, and Takeji Takui
DOI: 10.1021/acs.jpclett.2c02737
Publication date: November 29, 2022
Contact
Graduate School of Science
Dr. Kenji SUGISAKI
E-mail: sugisaki[at]omu.ac.jp
*Please change [at] to @.
Graduate School of Science
Professor Emeritus Takeji TAKUI
E-mail: takui[at]omu.ac.jp
*Please change [at] to @.
SDGs
